Modelling
-
MSIF - Poster Abstracts
Poster Abstracts
DEVELOPENT OF A COARSE-GRAINED POLYOXYETHYLENE GLYCOL NON-IONIC SURFACTANT MODEL USING THE SAFT-γ MIE FORCE-FIELD FOR MOLECULAR DYNAMICS SIMULATIONS - poster
Emma C L Richards, Erich A Müller and George Jackson
Department of Chemical Engineering, Imperial College London, London, UK
Robust coarse-grained (CG) models which exhibit both quantitative accuracy and representability are needed if one expects to obtain valuable insights to complex fluid systems. Within this study the SAFT-γ force-field methodology previously introduced in our group is employed to develop a CG surfactant model for use in the molecular simulation of non-ionic alkyl poly(oxyethylene) glycols (POE). In this approach, the effective Mie (generalized Lennard-Jones) interactions between the CG beads are estimated directly from target macroscopic thermodynamic properties with the aid of the molecular-based SAFT-γ Mie equation of state. Aqueous mixtures of alkyl poly(oxyethylene) glycol surfactants, CiEj , are considered as prototypical systems, where the interactions between the CG constitutive chemical moieties (alkyl, ether and hydroxyl groups), and their interactions with water are parameterised using appropriate experimental data for the vapour-liquid equilibria, liquid-liquid equilibria, enthalpy of mixing, and interfacial tension of selected pure components and mixtures. Here, a breakdown of the development and theory of the model will be discussed, as well as discussion of future applications such as the study of rheology and mesophase morphology.
SAFT γ-MIE COARSE-GRAINED FORCEFIELD FOR THE SIMULATION OF ANIONIC SURFACTANTS: PHASE BEHAVIOUR - poster
Matthias Kiesel, George Jackson, Erich A. Müller, Amparo Galindo
Department of Chemical Engineering, Centre for Process Systems Engineering, Institute for Molecular Science and Engineering, Imperial College London, London SW7 2AZ, U.KIn order to describe and predict the phase behaviour of amphiphilic molecules with standard molecular simulation, a balance must be made between an accurate description at the molecular level and computational cost. In previous works, forcefields based on the SAFT γ-Mie equation of state 1,2,3 have been derived. A major benefit of this top-down approach is the analytical parameterization, which facilitates the inclusion of a wide range of experimental data, delivering increased transferability and representability to the model. In this work, we extend our SAFT γ-Mie forcefield development workflow to develop models for charged species. Strong electrolytes are modelled as Mie segments with central point charges, interacting via a screened coulomb potential. Interactions between electrolytes and a water model2 are incorporated in a Mie potential, optimised to reproduce osmotic coefficients. The model can reproduce osmotic coefficients and densities of solvated electrolytes over the concentration and temperature range of interest.
1: Papaioannou et al., J. Chem. Phys. 140, 054107 (2014)
2: Lobanova et al., Mol. Phys., 113:9-10, 1228-1249 (2015)
3: Rahman et al., J. Phys. Chem. B 122, 9161−9177 (2018)FORCE FIELD ASSESSMENT IN THE MOLECULAR SIMULATION OF INTERFACIAL TENSIONS OF SURFACTANTS - poster
Harry Cárdenas1, Siti Fatihah Salleh2, Sara Shahruddin2, and Erich A. Müller1
1Chemical Engineering Department, Imperial College London, UK.
2Specialty Chemicals Technology, PETRONAS Research Sdn Bhd, MALAYSIA
With advancement in algorithms and computational power, it is now possible to practically model systems that closely resemble those studied in the laboratory using atomistic molecular dynamics simulations. However, the impressive visual graphics and the detailed molecular insight obtained from the calculations distract from the fact that these representations and their quantitative predictions are only as good as the quality of the underlying force fields used to describe the intermolecular interactions. Hence, it is critical to identify the right intermolecular potential with the appropriate balance of accuracy, transferability and representability. We present here an example to demonstrate the impact of force field selection for surfactant performance assessment.
The systems under study herein include alkyl polyglucosides (APG) molecules; biocompatible and degradable surfactants formed by a sugar-based hydrophilic head, and a hydrocarbon-based hydrophobic tail. Two different interfacial environments are considered: water/air interface and water/oil interface, where the oil is phase is represented by n-decane molecules. Our initial simulations show that the pcff+ water model induces a physically unrealistic behaviour for the surfactants at the interface, particularly noticeable at the water-air interface (Figure 1) which are improved by employing an older model, (labelled here as pcff). The work discusses these issues in depth and served to highlight the general question of how much confidence we can/should have on the predictions of molecular dynamics simulations and the importance of testing the validity of the potentials against experimental results.
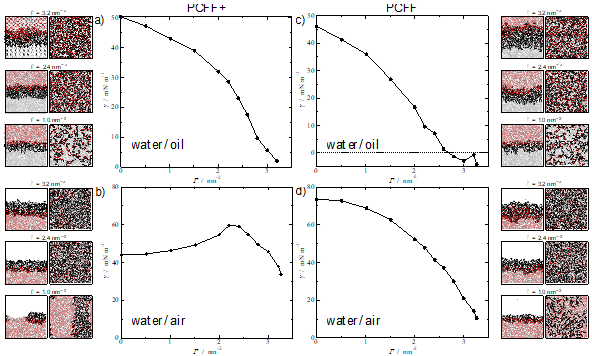
Figure 1. Interfacial tension at 298.15 K of: (top) APG12 at water/decane interface; (bottom) APG12 at water/air interface. The left figures correspond to the use of the pcff+ water model while those on the right with the older pcff water model. APG12 and n-decane molecules were modelled with pcff+ for all the systems. Snapshots for three different surface coverages values are added for each system, showing side and top view.
COMPUTER SIMULATIONS OF EPOXY BINDING ON IRON OXIDE SURFACES - poster
Charlie Wand1, Simon Gibbon2 and Flor Siperstein1
1 Department of Chemical Engineering and Analytical Science, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
2 AkzoNobel Research & Development, Northallerton, North Yorkshire, DL7 7BJ, UK
This email address is being protected from spambots. You need JavaScript enabled to view it. Epoxy resins are widely used in protective coatings due to their good heat and chemical resistance, favourable mechanical properties and good adhesion to a range of substrates. As such, epoxy resins have been formulated as a protective coating for a wide range of applications, from aerospace and marine applications through to nontoxic interior coatings in the food industry [1]. In all cases, the performance of the final solid-polymer system is dependent on the physicochemical properties of the interface and the interaction between the polymer and the solid substrate. However, experimental methods to characterize this interaction are limited and mostly deteriorative to the interface. Computer modelling provides a tool to investigate the surface-polymer interface at an atomistic level.
Here we perform atomistic molecular dynamics simulations to investigate the binding of a common component in epoxy resins, diglycidyl ether of bisphenol A (DGEBA), on Iron Oxide surfaces (Figure 1) and investigate the effect of number of repeat units in DGEBA on the binding energy (ΔEbinding) defined as;
ΔEbinding= Eadsorbate/surface-(Eadsorbate+Esurface )
Where Eadsorbate/surface is the energy of the adsorbed DGEBA on the surface and Eadsorbate, Esurface are the energy of the adsorbate and surface in vacuum respectively. In epoxy resin applications the composition of the solid substrate is highly varied, with pre-treatments and production processes leading to a non-uniform surface chemistry and roughness. To reflect this, we investigate two Iron Oxides surfaces, hematite (Fe2O3) and magnetite (Fe3O4). We find that binding is stronger for DGEBA on hematite than magnetite, in agreement with previous literature findings [2] and suggest causes of this trend based on the surface termination.

This work was done with support from the EPSRC Prosperity Partnership SusCORD (EP/S004963/1).
[1] Friedrich, Jörg. Metal-Polymer Systems: Interface Design and Chemical Bonding. John Wiley & Sons, 2017.
[2] Bahlakeh, Ghasem, et al J. Phys. Chem. C 120 20 (2016): 11014-11026.
LEVERAGING FIT TO PURPOSE DRUG RELEASE MODELS TO HELP WITH FORMULATION DEVELOPMENT AND SCALE-UP EFFORTS - poster
Ravichandra Palaparthi, PhD[a,b]
[a]Anagha Consultants, Hyderabad 500050 India, [b]Anagha Consultants LLC, Hockessin DE 19707, United States
This email address is being protected from spambots. You need JavaScript enabled to view it. The quality of a product produced from a process is dependent on how it is formulated (the ingredients, their composition), but also on the process conditions of production, and the characteristics of the equipment used. Understanding of the interaction of these different factors, in addition to how the ingredients of the formulation interact with each other is important to achieve a consistent product quality. This is especially true for the pharma and specialty materials industry, where consistently maintaining finished product quality is critical. Hence formulators need to account for these interactions early on in their formulation development efforts and ensure they go hand in hand with the process/scale-up ones. A combination of fit for purpose process and application specific models can be leveraged to get actionable insights into the necessary experimentation to meet drug product quality attributes.
This poster and video focus on an example case study in this direction from the pharma space of sustained release microspheres products. It shows how a model for drug release [1] can be leveraged to bring insights into necessary experimentation to troubleshoot issues with variability in finished product's drug release rates. Use of such customized application specific models along with process models can help provide predictability to (and address troubleshoot such issues during) scale-up. Such customized tools facilitate the scientists and engineers in the industry to effectively leverage the power of simulations in their routine needs.
1. Mehan, et al AIChE Annual Conference, 2016
-
MSIF20 - A Del Regno - Molecular modelling for discovery and optimisation of liquid formulations
Molecular modelling for discovery and optimisation of liquid formulations
Annalaura Del Regno, Stephan Köhler, Peter Müller and Eduard Schreiner
BASF SE, Materials Molecular Modeling, Ludwigshafen, GermanyIn a fast-growing economy, in which new products are brought to market every day, quick development of fine-tuned formulations is the key to success. To stay competitive in such a rapidly changing environment, BASF uses various digital technologies to speed up the development process and thus to support their customers and partners. Product development is an iterative process involving many steps and cycles. Our team develops and deploys computational methods to assist in the discovery and development of novel formulation products and the optimisation of existing ones. The goal of our efforts is to reduce the number of experiments and iteration cycles needed to bring a formulation from the lab-bench to the shelf. By utilizing the power of BASF’s Quriosity super computer and working closely with our experimental partners, we can accelerate the discovery process by (1) screening over hundreds of different formulations in a short period of time, (2) highlighting the most promising candidate, (3) understanding the microscopic interactions that make our products stable and (4) providing insight into their mechanism of action. Here, we will present an overview on how our parametrisation strategy, in combination with atomistic and coarse-grained modelling can be used to support the development of novel formulations in the home care, agriculture and oil sectors. We will show how these methods can be applied to study complex laundry formulations and determine washing performances, screen over different ingredients to improve the loading and emulsification behaviour of crop care formulations, and guide through the selection of different viscosity index improvers in the automotive sector.
-
MSIF20 - B OConchuir - The Role of Topology in Formulation Modelling and Simulation
The Role of Topology in Formulation Modelling and Simulation
Dr Breanndán Ó Conchúir
IBM ResearchIn this emerging era of data-driven material discovery, the application of mathematical methods is playing an increasingly important role in chemical formulation modelling and simulation. In particular, algorithms from the field of topology can be used to complement the scientific insights gained from molecular and mesoscopic simulations. In this talk, we will investigate this theme by examining two distinct problems in chemical formulation. In the first project1,2, we develop a topological algorithm to characterise ion-induced structural and kinetic transitions in micellar assemblies. This is important because small fluctuations in the complex morphology of micelles, produce drastic variations in the viscosity of liquid-based surfactant formulations. Meanwhile, in the second study3, we explore the role of dual-component chemical patterning on surfactant adsorption at the solid-liquid interface. We show how calibrating the topology of the hydrophobic-hydrophilic surface allows one to tune the spatial distribution of adsorbed molecules.
This work was supported by the STFC Hartree Centre’s Innovation Return on Research programme, funded by the Department for Business, Energy & Industrial Strategy
1) Garnder et al. SIAM News, (2020)
2) Conchuir et al. J. Chem. Theory Comput. 16, 7, 4588 (2020)
3) Klebes et al. J. Chem. Theory Comput. (2020) - Accepted
-
MSIF20 - C Adjiman - The role of model-based optimisation in designing formulations
The role of model-based optimisation in designing formulations
Claire Adjiman
Department of Chemical Engineering, Imperial College LondonThe need for extensive experimental campaigns to design new formulations can lead to high development costs and, in cases where new materials are involved (e.g., a new active pharmaceutical ingredient) and there is therefore limited supply. Planning an experimental campaign, using a method such as factorial design for instance, is especially difficult when some of the factors include discrete options, such as a choice of polymers, surfactants or solvents.
In this talk, we explore ways in which model-based optimisation can facilitate the design of formulations. We focus in particular on approaches that enable different ingredients to be selected for the final formulation and that lead to a list of promising designs to be tested experimentally.
We focus on the design of experiments to improve performance. We discuss the use of computer-aided molecular design to guide the search for better performing molecules and formulations. This requires the integration of structure-property models and property-performance models within an optimisation framework and leads to a prioritised list of candidate designs that can be tested experimentally. When the necessary models are not available, we show how physical or virtual experiments can be used to build surrogate models. These concepts are illustrated through examples from the design of reaction solvents and drug delivery systems.
-
MSIF20 - C Fonte - Modelling and simulation of application involving the dispensing of rheologically complex liquids
Modelling and simulation of applications involving the dispensing of rheologically complex liquids
Claudio P. Fonte
Department of Chemical Engineering and Analytical Science, The University of ManchesterThe ejection of rheologically complex liquids through round nozzles is a commonplace process in many industrial applications from inkjet printing and additive manufacture, to the dispensing of pharmaceutical and personal care products. Well-controlled dispensing of accurate volumes of these liquids is crucial in all of these applications. However, while the task may seem trivial, Non-Newtonian fluid properties, such as shear thinning and viscoelasticity, can often produce unexpected behaviours and challenges.
In this work, we show how numerical methods can provide reasonable predictions of how a particular liquid formulation will perform when ejected through a nozzle, by knowing its physical properties and rheological properties. We use Computational Fluid Dynamics to study the dynamics of a single pulse of inelastic and elastic fluids from round capillary tubes. More specifically, we observe how changes to the duration of the pulse and its velocity have an impact on the observed flow regimes.
From our results, we establish three main regimes: no drop detachment; single droplet formation; and satellite droplet formation. We find that viscoelasticity plays a significant role in the process of drop detachment by delaying breakup. We also find that changing the duration of the flow pulse can result in dramatically different regimes, even when the same jetted volume is kept, which can have a crucial role on the control of flow regimes for a wide range of industrial applications.
-
MSIF20 - C Wand - Computer simulations of epoxy binding on iron oxide surfaces
COMPUTER SIMULATIONS OF EPOXY BINDING ON IRON OXIDE SURFACES - poster
Charlie Wand1, Simon Gibbon2 and Flor Siperstein1
1 Department of Chemical Engineering and Analytical Science, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
2 AkzoNobel Research & Development, Northallerton, North Yorkshire, DL7 7BJ, UK
This email address is being protected from spambots. You need JavaScript enabled to view it. Epoxy resins are widely used in protective coatings due to their good heat and chemical resistance, favourable mechanical properties and good adhesion to a range of substrates. As such, epoxy resins have been formulated as a protective coating for a wide range of applications, from aerospace and marine applications through to nontoxic interior coatings in the food industry [1]. In all cases, the performance of the final solid-polymer system is dependent on the physicochemical properties of the interface and the interaction between the polymer and the solid substrate. However, experimental methods to characterize this interaction are limited and mostly deteriorative to the interface. Computer modelling provides a tool to investigate the surface-polymer interface at an atomistic level.
Here we perform atomistic molecular dynamics simulations to investigate the binding of a common component in epoxy resins, diglycidyl ether of bisphenol A (DGEBA), on Iron Oxide surfaces (Figure 1) and investigate the effect of number of repeat units in DGEBA on the binding energy (ΔEbinding) defined as;
ΔEbinding= Eadsorbate/surface-(Eadsorbate+Esurface )
Where Eadsorbate/surface is the energy of the adsorbed DGEBA on the surface and Eadsorbate, Esurface are the energy of the adsorbate and surface in vacuum respectively. In epoxy resin applications the composition of the solid substrate is highly varied, with pre-treatments and production processes leading to a non-uniform surface chemistry and roughness. To reflect this, we investigate two Iron Oxides surfaces, hematite (Fe2O3) and magnetite (Fe3O4). We find that binding is stronger for DGEBA on hematite than magnetite, in agreement with previous literature findings [2] and suggest causes of this trend based on the surface termination.
This work was done with support from the EPSRC Prosperity Partnership SusCORD (EP/S004963/1).
[1] Friedrich, Jörg. Metal-Polymer Systems: Interface Design and Chemical Bonding. John Wiley & Sons, 2017.
[2] Bahlakeh, Ghasem, et al J. Phys. Chem. C 120 20 (2016): 11014-11026.
-
MSIF20 - E Richards - Development of a coarse-grained polyoxyethylene glycol non-ionic surfacant model using the saft-g Mie force-field for molecular dynamics simulations
DEVELOPENT OF A COARSE-GRAINED POLYOXYETHYLENE GLYCOL NON-IONIC SURFACTANT MODEL USING THE SAFT-γ MIE FORCE-FIELD FOR MOLECULAR DYNAMICS SIMULATIONS - poster
Emma C L Richards, Erich A Müller and George Jackson
Department of Chemical Engineering, Imperial College London, London, UK
Robust coarse-grained (CG) models which exhibit both quantitative accuracy and representability are needed if one expects to obtain valuable insights to complex fluid systems. Within this study the SAFT-γ force-field methodology previously introduced in our group is employed to develop a CG surfactant model for use in the molecular simulation of non-ionic alkyl poly(oxyethylene) glycols (POE). In this approach, the effective Mie (generalized Lennard-Jones) interactions between the CG beads are estimated directly from target macroscopic thermodynamic properties with the aid of the molecular-based SAFT-γ Mie equation of state. Aqueous mixtures of alkyl poly(oxyethylene) glycol surfactants, CiEj , are considered as prototypical systems, where the interactions between the CG constitutive chemical moieties (alkyl, ether and hydroxyl groups), and their interactions with water are parameterised using appropriate experimental data for the vapour-liquid equilibria, liquid-liquid equilibria, enthalpy of mixing, and interfacial tension of selected pure components and mixtures. Here, a breakdown of the development and theory of the model will be discussed, as well as discussion of future applications such as the study of rheology and mesophase morphology.
-
MSIF20 - GM Kontogeorgis - Computer-Aided Design of Paints and Coatings - A review and recent applications
Computer-Aided Design of Paints and Coatings – A review and recent applications
Georgios M. Kontogeorgis*, Spardha Jhamb, Xiaodong Liang, Kim Dam-Johansen
CERE and CoaST, Department of Chemical and Biochemical Engineering, Technical University of Denmark, Building 229, Søltofts Plads 229, DK – 2800, Kgs. Lyngby, Denmark
*Corresponding author
Email address:This email address is being protected from spambots. You need JavaScript enabled to view it. Computer-aided design (CAD) is still in an early stage in the paints and coatings industrial sector and its potential is yet to be utilised to the maximum. Nevertheless, Computer-aided tools offer many possibilities in the design of paints and coatings. Significant advances have been made, involving also the use of thermodynamic and other property models for the study and theoretical formulation of these products. Algorithms and tools based on such models enable the formulation chemist to speed up the design process, by allowing them to focus their experimental efforts on a selected number of reliable constituents for the coating formulation. The final validation should still be done using experiments.
In this presentation, we first briefly present some literature studies in the field of CAD for paints and coatings, which are based on physicochemical property models or on machine learning algorithms and high-throughput experimentation.
Next, we turn our focus on the selection of solvents and calculation of their composition in the final product, which is a critical step in coating formulation design. This task can be greatly facilitated by computer-aided methods if the necessary thermodynamic and group contribution property models along with chemical property data are available for the ingredients under consideration. We will show that the computer-aided stage can be used to speed-up the solvent selection and design process, efficiently utilize experimental resources and serve as a guide for the formulation chemist.
We will particularly illustrate in this work an adaption of the generic Computer-Aided Product Design (CAPD) methodology for organic coating formulations with focus on the design of solvents for coatings. The applicability of this framework will be tested via case studies with industrial interest which involve a variety of pigments, solvents and polymers.
The scope of using the computer-aided design algorithms is limited by the availability, reliability and accuracy of the models employed to predict the target properties. Limitations of the current framework and future directions will also be outlined.
-
MSIF20 - H Cardenas - Force field assessment in the molecular simulation of interfacial tensions of surfactants
FORCE FIELD ASSESSMENT IN THE MOLECULAR SIMULATION OF INTERFACIAL TENSIONS OF SURFACTANTS - poster
Harry Cárdenas1, Siti Fatihah Salleh2, Sara Shahruddin2, and Erich A. Müller1
1Chemical Engineering Department, Imperial College London, UK.
2Specialty Chemicals Technology, PETRONAS Research Sdn Bhd, MALAYSIA
With advancement in algorithms and computational power, it is now possible to practically model systems that closely resemble those studied in the laboratory using atomistic molecular dynamics simulations. However, the impressive visual graphics and the detailed molecular insight obtained from the calculations distract from the fact that these representations and their quantitative predictions are only as good as the quality of the underlying force fields used to describe the intermolecular interactions. Hence, it is critical to identify the right intermolecular potential with the appropriate balance of accuracy, transferability and representability. We present here an example to demonstrate the impact of force field selection for surfactant performance assessment.
The systems under study herein include alkyl polyglucosides (APG) molecules; biocompatible and degradable surfactants formed by a sugar-based hydrophilic head, and a hydrocarbon-based hydrophobic tail. Two different interfacial environments are considered: water/air interface and water/oil interface, where the oil is phase is represented by n-decane molecules. Our initial simulations show that the pcff+ water model induces a physically unrealistic behaviour for the surfactants at the interface, particularly noticeable at the water-air interface (Figure 1) which are improved by employing an older model, (labelled here as pcff). The work discusses these issues in depth and served to highlight the general question of how much confidence we can/should have on the predictions of molecular dynamics simulations and the importance of testing the validity of the potentials against experimental results.
Figure 1. Interfacial tension at 298.15 K of: (top) APG12 at water/decane interface; (bottom) APG12 at water/air interface. The left figures correspond to the use of the pcff+ water model while those on the right with the older pcff water model. APG12 and n-decane molecules were modelled with pcff+ for all the systems. Snapshots for three different surface coverages values are added for each system, showing side and top view.
-
MSIF20 - L Mazzei - Modelling and Simulation Applied to Sonocrystallization in Continuous-Flow Millichannel Contractors
Modelling and Simulation Applied to Sonocrystallization in Continuous-Flow Millichannel Contactors
Luca Mazzei
Department of Chemical Engineering, University College London, UKCrystallization is widely employed in pharmaceuticals manufacturing; over 90% of all pharmaceutical products contain drugs in particulate, and usually crystalline, form. Typically, crystallizers are standard agitated vessels, and seeding is used to direct and control the nucleation process. Major challenges persist in conventional crystallizers regarding process controllability and product reproducibility owing to the complexity of the crystallization process, which involves several rapid, concurrent and closely interacting phenomena (e.g. nucleation and growth); this leads to wide particle size distributions and substantial batch-to-batch product variability, resulting in pharmaceutical formulation problems, such as bioavailability and drug stability. To overcome these challenges, one should design new-generation crystallizers able to deliver a step-change in particle production technology.
Continuous-flow processing and ultrasound have the potential to overcome these issues. Continuous crystallizers can operate in steady-state conditions and, if their size is small, intensify heat and mass transfer, thus improving process control and product reproducibility; ultrasound induce cavitation and acoustic streaming, allowing us to trigger nucleation on demand and alter the flow patterns and mixing within the unit. But to harness this potential, one should fully understand the effect of each additional degree of freedom introduced by ultrasound and be able to characterize the ultrasonic field and flow patterns in the crystallizer. Modelling and simulations can help understand better sonocrystallization and develop, design and optimize sonocrystallizers.
In this presentation, we investigate continuous-flow sonocrystallization of adipic acid in milli-channels. We use modelling and simulations to characterize cavitation and acoustic streaming in the crystallizer, relating them to nucleation, flow pattern, residence time distribution (RTD), mixing and crystallization performance. We show that inertial cavitation is closely related to nucleation and acoustic streaming can strongly affect the performance and behaviour of the crystallizer. In particular, a small increase in the capillary diameter can invert important trends, such as the mean crystal size against the sonication time: while in a 1.55 mm I.D. capillary the former increases with the latter, in a 3.2 mm I.D. capillary the opposite happens. This difference highlights the importance of acoustic streaming and shows that modelling and simulations are important design and optimization tools.
-
MSIF20 - M Kiesel - Saft g-Mie coarse-grained forcefield for the simulation of anionic surfactants: phase behaviour
SAFT γ-MIE COARSE-GRAINED FORCEFIELD FOR THE SIMULATION OF ANIONIC SURFACTANTS: PHASE BEHAVIOUR - poster
Matthias Kiesel, George Jackson, Erich A. Müller, Amparo Galindo
Department of Chemical Engineering, Centre for Process Systems Engineering, Institute for Molecular Science and Engineering, Imperial College London, London SW7 2AZ, U.KIn order to describe and predict the phase behaviour of amphiphilic molecules with standard molecular simulation, a balance must be made between an accurate description at the molecular level and computational cost. In previous works, forcefields based on the SAFT γ-Mie equation of state 1,2,3 have been derived. A major benefit of this top-down approach is the analytical parameterization, which facilitates the inclusion of a wide range of experimental data, delivering increased transferability and representability to the model. In this work, we extend our SAFT γ-Mie forcefield development workflow to develop models for charged species. Strong electrolytes are modelled as Mie segments with central point charges, interacting via a screened coulomb potential. Interactions between electrolytes and a water model2 are incorporated in a Mie potential, optimised to reproduce osmotic coefficients. The model can reproduce osmotic coefficients and densities of solvated electrolytes over the concentration and temperature range of interest.
1: Papaioannou et al., J. Chem. Phys. 140, 054107 (2014)
2: Lobanova et al., Mol. Phys., 113:9-10, 1228-1249 (2015)
3: Rahman et al., J. Phys. Chem. B 122, 9161−9177 (2018) -
MSIF20 - M Sarwar - Computational Chemistry: An Industrial Perspective
Computational Chemistry : An Industrial Perspective
Misbah Sarwar
Johnson MattheyThis talk will give an overview of how multiscale modelling approaches are used to understand formulations used in automotive catalysis to reduce emissions from gasoline and diesel vehicles. Cu zeolites are used to reduce NOx from diesel emissions and the talk will show how DFT simulations are used to understand Cu location in zeolites and interaction with SCR reactants such as NO and NH3. Moving up the length scales the talk will then describe how forcefield based MD simulations are used to understand diffusion of NH3 in different zeolite frameworks with validation from QENS measurements. Finally, current work and future directions on applying mesoscale modelling techniques to model properties of washcoat formulations will be discussed.
-
MSIF20 - V Ginzburg - Using First-Principles Multi-Scale Modelling to Design Rheology Modifiers for Waterborne Paints
Using First-Principles Multi-Scale Modeling to Design Rheology Modifiers for Waterborne Paints
Valeriy Ginzburg
Dow Inc. (retired)Modern paints are complex mixtures comprised of multiple components such as water, pigments, latex binders, dispersants, rheology modifiers or thickeners, and others. This presentation is specifically devoted to the use of modeling to understand the rheology and colloidal stability of a specific system -- hydrophobically ethoxylated urethanes (HEUR) thickeners in waterborne paints. Since their invention in 1970s, HEURs have been actively used as rheology modifiers for paints. Thermodynamic and rheological behavior of HEUR molecules in aqueous solutions is now very well understood and is based on the concept of transient network (TN), where the association of hydrophobic groups into networks of flower micelles causes viscosity to increase dramatically as function of polymer concentration. The behavior of complex mixtures containing water, HEUR, and latex (“binder”) particles, however, is understood less well, even though it has utmost importance in the paint formulation design. To model the colloidal stability and rheology of these systems, Dow partnered with the University of Michigan research team led by Prof. Ronald Larson. We developed a multi-scale modeling framework in which the molecular details of the HEUR polymers are used to describe their adsorption onto the binder particles and ultimately the viscoelastic behavior of the overall binder/HEUR aqueous dispersion. The model is able to qualitatively describe many important features of the water/latex/HEUR mixtures. The proposed approach could potentially lead to the design of new HEUR structures with improved rheological performance.
-
MSIF20 - Z Alam - Virtual Design of Laundry Formulations
Virtual Design of Laundry Formulations
Zayeed Alam
Director, Data & Modeling Sciences, Corporate Functions, Procter & Gamble
There are a range of technical and complexity challenges involved in designing a laundry detergent. Zayeed will touch on each of these and illustrate how at P&G his team have to date partnered externally to develop and leverage a range of state of the art Modeling & Simulation tools to tackle these challenges and where the future opportunities lie.
-
MSIF20- - R Palaparthi - Leveraging fit to purpose drug release models to help with formulation devleopment and scale-up efforts
LEVERAGING FIT TO PURPOSE DRUG RELEASE MODELS TO HELP WITH FORMULATION DEVELOPMENT AND SCALE-UP EFFORTS - poster
Ravichandra Palaparthi, PhD[a,b]
[a]Anagha Consultants, Hyderabad 500050 India, [b]Anagha Consultants LLC, Hockessin DE 19707, United States
This email address is being protected from spambots. You need JavaScript enabled to view it. The quality of a product produced from a process is dependent on how it is formulated (the ingredients, their composition), but also on the process conditions of production, and the characteristics of the equipment used. Understanding of the interaction of these different factors, in addition to how the ingredients of the formulation interact with each other is important to achieve a consistent product quality. This is especially true for the pharma and specialty materials industry, where consistently maintaining finished product quality is critical. Hence formulators need to account for these interactions early on in their formulation development efforts and ensure they go hand in hand with the process/scale-up ones. A combination of fit for purpose process and application specific models can be leveraged to get actionable insights into the necessary experimentation to meet drug product quality attributes.
This poster and video focus on an example case study in this direction from the pharma space of sustained release microspheres products. It shows how a model for drug release [1] can be leveraged to bring insights into necessary experimentation to troubleshoot issues with variability in finished product's drug release rates. Use of such customized application specific models along with process models can help provide predictability to (and address troubleshoot such issues during) scale-up. Such customized tools facilitate the scientists and engineers in the industry to effectively leverage the power of simulations in their routine needs.
1. Mehan, et al AIChE Annual Conference, 2016